[et_pb_section fb_built=”1″ _builder_version=”3.22″ global_colors_info=”{}”][et_pb_row _builder_version=”3.25″ background_size=”initial” background_position=”top_left” background_repeat=”repeat” global_colors_info=”{}”][et_pb_column type=”4_4″ _builder_version=”3.25″ custom_padding=”|||” global_colors_info=”{}” custom_padding__hover=”|||”][et_pb_text _builder_version=”3.27.4″ background_size=”initial” background_position=”top_left” background_repeat=”repeat” global_colors_info=”{}”]
- برای بهره مندی هرچه بیشتر و فهم صحیح تر درباره موضوع زیر، نیاز است تا با کلمات ذیل آشنا شوید:
FDA
FD&C
FDCA
سازمان ملی فرمولاسیون آمریکا(official National Formulary )
(o)520
بخش (h)201
تولیدکنندگان دستگاههای پزشکی در ایالات متحده به این نیاز دارند
که محصولات خود را در سازمان غذا و دارو (FDA) ثبت کنند. در ادامه نیز می بایست دستگاههای
مشخصی را برای بررسی نیازمندیهای سختگیرانهای که شامل آزمایشهای بالینی میشوند
، ارسال میکند. از آنجایی که بسیاری از قانونگذاران در دیگر کشورها براحتی برای دستگاههایی
که قبلأ توسط FDA تأیید شده بود، مجوز میدهند لذا شرکتهایی که سودای جهانی شدن دارند
بیشتر به دنبال این هستند که با دسترسی به بازار ایالات متحده شروع کنند. FDA دستگاههای
پزشکی را بر اساس استفادههای مورد انتظار و هدف مورد استفاده، با استفاده از یک مقیاس،
دستهبندی میکنند. این دسته بندی بر اساس ریسک محصول و بر اساس سه سطح از ریسک (گروه
های I، II و III) تعریف می شود. آوردن یک دستگاه جدید به بازار به برنامهریزی از قبل
برای کارهای ضروری برای بدست آوردن و قطعی کردن تأیید FDA نیاز دارند. یک اشتباه جدی
میتواند سبب شود که FDA درخواست شما را رد کند یا محصول را از بازار خارج کند. برای
جلوگیری از شکست، گام اول شما باید یادگیری درباره این باشد که فرایند ثبت چگونه کار
میکند و شرکت شما در کجا به کمک نیاز دارد.
FDA ایالات متحده چه چیزی را بعنوان دستگاه پزشکی در نظر میگیرد؟
در ابتدای امر واژه تجهیزات پزشکی ممکن است دستگاههای سطح بالای
فناورانه و ایمپلنت های کاشتننی را به ذهن القا کند؛ درحالیکه تجهیزات پزشکی شامل دستگاه
های بسیار کوچک و رایج نظیر ماسک و دماسنج جیوه ای را نیز شامل می شود . FDA
ایالات متحده تجهیزات پزشکی را به شیوهای شبیه به نحوه مدیریت داروها قانونگذاری میکند.
با این حال، قانونگذاری تجهیزات پزشکی بعدها به میزان زیادی توسعه یافته است. در 1976،
کنگره اصلاحیهای را به قانون فدرال غذا، دارو و لوازم آرایشی اضافه کرد (FD&C یا FDCA)
که بخش (h)201 آن یک وسیله پزشکی را به این صورت تعریف میکند:
یک ابزار، وسیله، وسیله کاشتنی ، ماشین، تمهید، پیوند، یک معرف آزمایشگاهی، یا دیگر موارد مشابه یا مرتبط شامل یک بخش جزئی، یا ضمیمهای که ویژگیهای زیر را دارد:
- در سازمان ملی فرمولاسیون آمریکا(official National Formulary
) یا دایرهالمعارف دارویی ایالات متحده(فارماکوپه)، یا هر ضمیمه
آنها ثبت شده باشد. برای استفاده در تشخیص بیماری یا دیگر شرایط، یا در درمان،
تسکین، بهبودی یا پیشگیری از بیماری، در انسان یا دیگر حیوانات استفاده شود.
به منظور اثرگذاری بر ساختار یا هرگونه عملکرد بدن انسان یا دیگر حیوانات طراحی شده باشد، و این کار با اهداف اولیه آن از طریق عمل شیمیایی در داخل یا بروی بدن انسان یا دیگر حیوانات بدست نیاید و وابسته به این نباشد که برای دستیابی به هدف اولیه که بدان منظور طراحی شده است، متابولیسم بدن را تغییر دهد. واژه “تجهیز” شامل فعالیتهای نرم افزاری مجزا شده برای دنبالکنندگان در بخش (o) 520 نمیباشد
درحالیکه انواع معینی از نرم افزارها از این تعریف توسط بخش (o)520 از FD&C مستثنی میشوند، بسیاری از اجزای نرم افزاری تجهیزات پزشکی و همچنین نرم افزارهایی که به تنهایی کار میکنند، بعنوان وسایل پزشکی توسط FDA دستهبندی میشوند و باید برای تأیید شدن ثبت شوند.
تفاوتها بین دستههای مختلف تجهیزات پزشکی در FDA چیست؟ FDA سه دسته متفاوت دستگاههای پزشکی بر پایه ریسک احتمالی را به رسمیت میشناسد، درحالیه مراجع قانونی در کشورهایی مانند کانادا و ژاپن از چهار دسته استفاده میکنند و اتحادیه اروپا نیز دسته بندی خود را دارد
وسعت تجهیزات پزشکی در ایالات متحده، بیان کننده این موضوع است که نیاز به دسترسی به بازار میتواند برای محصولات متفاوت باشد و در عین حال در یک کلاس خطر( Risk Class) قرار گیرند. به بیانی بهتر می توان گفت که دستهبندیهای عمومی یک وسیله نمیتواند بعنوان یک نقشه راه کامل برای ثبت و تأیید آنها در FDA نظرگرفته شود.
تجهیزات با کلاس خطر I، بعنوان دستگاههایی با ریسک پایین و متشکل از یک طرح ساده بدون قطعات حرکتکننده در نظر گرفته میشود؛ اینها نیازی ندارند که مستقیمأ زندگی یک فرد را نجات یا آن را حفظ کنند. برای دستگاههای گروه I شرکت تولید کننده بایستی که کنترلهای عمومی(general controls) را رعایت نماید. مثالهایی از این گروه شامل بانداژهای چسبنده، اسکالپل، و گوشی پزشکی میباشد. بسیاری از وسیلههای گروه I میتواند در FDA بدون نیاز به بدست آوردن مجوز ثبت شوند، اما برخی از آنها به اعلامیههای پیش بازاری 510(k) نیاز دارند(اصطلاحا به این موضوع 510k Notification می گویند).
تجهیزات با کلاس خطر II ، ریسک احتمالی متوسطی دارند، و غالبأ همراه با طراحیهای پیچیده هستند، اما عدم عملکرد آنها احتمالأ خطر آنی یا آسیب جدی یا مرگ را به دنبال ندارد. کنترلهای عمومی(general controls) برای تجهیزات با کلاس خطر II ناکافی هستند. این تجهیزات نیاز به کنترلهای ویژهای (special controls) از قبیل برچسبگذاری، استانداردهای عملکردی، و نظارتهای بعد از بازار (PMS – post market surveillance) دارند. از این گروه تجهیزات پزشکی می توان به آندوسکوپ، ویلچرهای برقی، سرنگها و ایمپلنت پیوند مفصلی اشاره کرد. برخی از تجهیزات با کلاس خطر II تنها نیاز به ثبت شدن دارند، اما بسیاری از آنها به مجوز نیاز دارند( در سازمان FDA به این موضوع اصطلاحا Clearance میگویند). مسیر اصلی به بازار برای وسیلههای این گروه ” اعلامیههای پیش از عرضه به بازار” است. در واقع تولید کننده تجهیزات پزشکی با کلاس خطر IIبایستی که سه آیتم زیر را رعایت نماید:
1- کنترل های عمومی
2- کنترل های ویژه
3- اعلامیههای پیش از عرضه به بازار(510(k) Premarket Notification )
تجهیزات پزشکی با کلاس خطر III ریسک بالایی دارند و میتوانند منجر به مشکلات پزشکی جدی از جمله مرگ در صورت عملکرد ناصحیح شوند. تجهیزات پزشکی با کلاس خطر III به این نیاز دارند که فرایند تأیید پیش از عرضه به بازار (PMA- Premarket Approval ) را سپری کنند، مگر اینکه ثابت شود اینها اساسأ هم ارز وسیلههای تأیید شده موجود در بازار هستند.
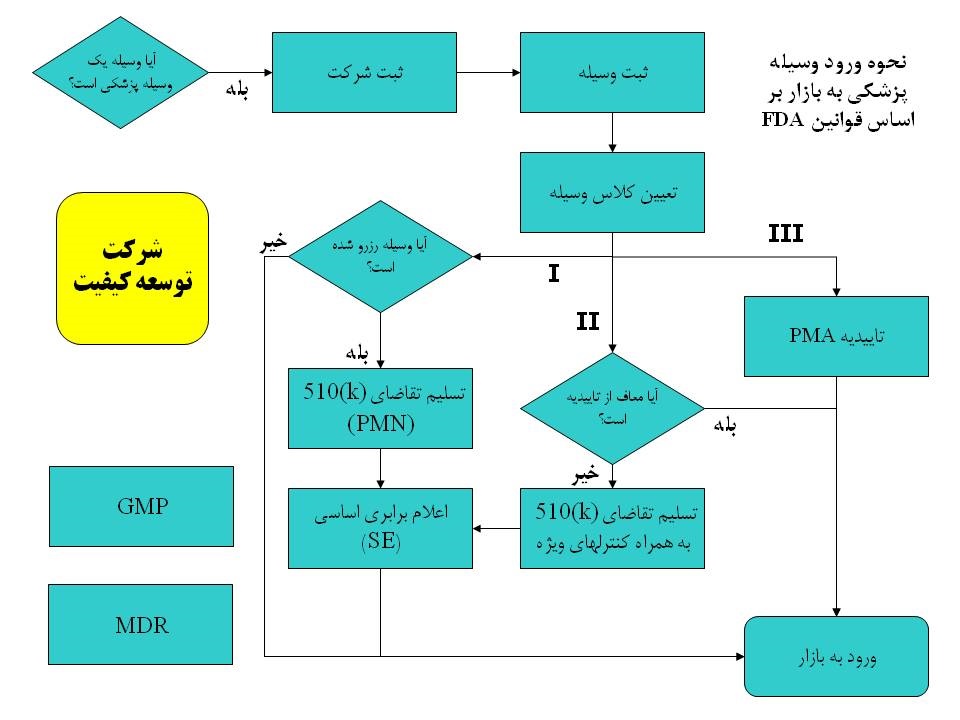
بعد از اینجا به کجا باید بروم؟
در حالیکه تعدادی ملاحظات اضافی وجود دارد که سازندگان تجهیزات پزشکی باید در نظر داشته باشند (برای مثال قابلیت استفاده داشتن، سیستم مدیریت کیفیت، طراحی یک پاسخ رسمی FDA به آن)، غیرممکن است که یک برنامه کامل برای دسترسی به بازار تا قبل از اطمینان از دستهبندی وسیله وجود داشته باشد. به انجام رسانیدن این گامهای ابتدایی در فرایند تأیید FDA ، به شکل مناسبی میتواند شما را در تولید و عرضه سریع محصول به بازار و عدم نیاز به شروع دوباره یاری دهد.
[/et_pb_text][/et_pb_column][/et_pb_row][/et_pb_section]